Genetic Sequencing
It took thirteen years to first sequence our DNA. Thanks to bioinformatics, we can now do it in a week.

It took thirteen years to first sequence our DNA. Thanks to bioinformatics, we can now do it in a week.
Imagine you have muscular dystrophy running through your family’s history. This is caused by genetic alterations or mutations — small changes in the genes that make up your body and dictate how your cells grow; changes that modify your body’s blueprint, so to speak, whether for the better or for the worse.
In this case, it’s definitely for the worse.
Muscular dystrophy is when those genetic mutations cause severe muscle weakness. Everything seems fine to start with, but, over time, they start getting weaker and weaker. What started out as a minor issue becomes a big deal, and more often than not, people with muscular dystrophy end up in a wheelchair because their legs can’t support them anymore.
Because muscular dystrophy is a genetic condition, there are no outward symptoms to see that something’s going wrong. No coughing, wheezing, or fevers here; nothing that can be checked with a thermometer or some other simple tool. Did you think the coronavirus was bad? This is even worse, because there’s mostly nothing — and then a sudden degeneration of the muscles when you least expect it. And no way of knowing beforehand.
No way, that is, until now.
Muscular dystrophy is one of those things that literally ‘runs in the family’. If you or a close relative have it, chances are your cousins and children will too.
But you have no way of knowing that, because symptoms are often delayed and take several years to physically show. Having been there all those years, however, the genetic condition will have already done its damage, at which point treatment is seldom easy.
Now we’re stumped. The family is running out of options, because they have the threat of muscular dystrophy hanging over them all their lives, and no way of knowing whether they’re worrying too much or too little. It seems like a dead-end, right?
Well, not quite, because there’s a special secret for these situations called bioinformatics!
If you think of bioinformatics as a fancy hi-tech term involving lots of jargon and software and computers, then you’re pretty close to the mark. What may surprise you, though, is that the term ‘bioinformatics’ is far from new. In fact, the word has been around right from the 1980s.
So what is bioinformatics, exactly? The brief version is that it’s the usage of software tools to understand and interpret biological and genomic data. Bioinformatics is what allows your DNA to be sequenced — your blueprint to be read, in other words — but, more importantly, it lets you compare your DNA with that of a healthy individual and monitor any changes. If a specialist detects a change linked to muscular dystrophy, for example, then maybe it’s time to be careful and run some followup tests.
If you want to go deeply into the history of bioinformatics, you’ll have to go back to the 1970s when computers were rare and expensive, and DNA sequencing wasn’t a thing. At that point, scientists were focusing more on protein biochemistry — that is, looking at the proteins that are made following the DNA’s instructions, because they didn’t have the technology to look at DNA itself.
The term ‘bioinformatics’ was coined by scientists Paulien Hogeweg and Ben Hesper in 1970, who described it as “the study of informatic processes in biotic systems.” Compared to the current definition, Hogeweg and Hesper were correct in saying that bioinformatics extracts information from a biological system.
Though the term itself was coined in 1970, notable scientists have used and investigated bioinformatics since the 1950s — they just didn’t call it that.
Margaret Dayhoff was a professor at Georgetown University Medical Center, but also a physical chemist and a notable pioneer in bioinformatics. She took protein sequences published in either research articles or books, and — thanks in part to her exposure to the IBM 7094 in the late 1950s — used a computer model to search for significant differences between the sequences.
Dayhoff also managed to find out about the proteins’ evolutionary sequences using sequencing alignment. She would rearrange the DNA, RNA, or a protein sequence, in order to spot the similarities or differences between two different sets of information.
In 1990, scientists around the world teamed up on what has been dubbed “one of the great feats of exploration in history”.
The plan was to go through the entire DNA of a human being, and map down all the genes — together known as the ‘genome’ — of the human species. Of course, they would look at one specific human, but the idea was to get the big picture of how all humans were coded. The project gave tons of valuable insight into human genetics and helped geneticists to better understand how the genome mutated and changed over time.
The Human Genome Project involved six countries and took 13 years to complete, at the cost of what was then 2.7 billion dollars. And that was just to sequence the genome of one person. Today, companies like 23andMe let individuals sequence their own genomes at the drop of a hat.
So what changed between 1991 and today? The answer lies partly in machinery. Manually sequencing anyone’s DNA would take years of gruelling work, not to mention higher expenses. With the help of advancing machinery, the human genome can now be sequenced for roughly $100 — a long cry from the Human Genome Project.
Before we look into the recent technologies used to sequence DNA, we first need to understand the techniques used by these devices. In bioinformatics, there are two types of sequencing methods: Sanger sequencing and next-generation sequencing.
In any sequencing method, we first need to identify an area of interest, which we’ll try to sequence. DNA is made up of small ‘nucleotides’ which act as the alphabets of the DNA. But these alphabets come in only four varieties: adenine, cytosene, guanine and thymine— or A, T, C and G for short. Our job is to read these alphabets and record them on the computer.
In Sanger sequencing, we copy the target DNA multiple times, creating multiple fragments of different lengths.
In Sanger sequencing, we copy
fragments of different
target DNA multiple
sequencing, we copy
the target of different lengths.
multiple times, creating fragmentsThese fragments will later be pieced together, in the same way that a set of photos comes together to form a panorama.
How do we decide where the fragments end? Well, when an oxygen atom is removed from a ordinary nucleotide, it creates a ‘dideoxy nucleotide’. No more nucleotides can be added on to it, so that’s where the DNA copying stalls. You can think of them as the ‘full stop’ of a sentence.
So we add the sample to a tube along with ‘DNA polymerase’ — the enzyme that actually copies DNA strands — and random nucleotide pieces for the polymerase to work on. Also in the tube are bits of ‘primer’: a small bit of DNA for the polymerase to start its work on. The ‘full stop’ dideoxy nucleotides are included too, but in much smaller amounts than the ordinary ones.
There’s one problem: we can’t ‘read’ nucleotides directly. What we can read are special nucleotides which are dyed in fluorescent colours especially for this purpose — which is exactly what the dideoxy nucleotides are!
And there we have the whole setup. Raise the temperature to separate the DNA strands, cool it to let DNA polymerase bind to the strands, and then raise it again to let DNA polymerase begin its copy-paste job. At some random point, the enzyme will hit a dideoxy nucleotide and stop, and you’ll end up with a random-length fragment.
Repeat, and repeat again, till you’ve got a lot of fragments of different lengths; each end will be labeled with dyes that show their final nucleotide. And then, it’s time for stage two.
Run these fragments through a gel-filled glass column, in a process known as capillary electrophoresis. An electrical field is added to the glass column so that the negatively-charged fragments migrate to the positive end, and vice versa.
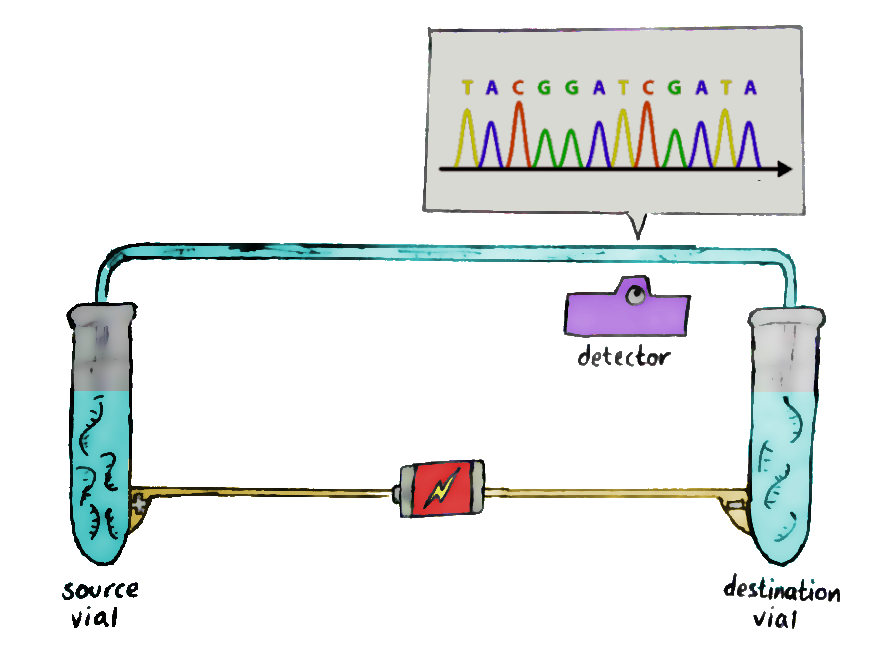
The short fragments move more quickly through the gel than the long ones, and so, at the end of the column, the shorter ones get detected first. This allows the sequence of the original DNA to be retained — and recorded by the laser waiting at the end.The data recorded by the laser shows successive peaks in what is known as a ‘chromatogram’, and that’s where you read the DNA from!
As science has progressed, people have come up with a better sequencing method: next-generation sequencing, or NGS, which helps analyze several million DNA sequences at once. Conceptually, next-generation sequencing is kind of like running a very large number of tiny Sanger sequencing reactions in parallel.
Although it bears resemblance to the Sanger method, there are some differences that make NGS more cost-efficient and time-saving. Instead of analysing sequences one by one, it processes many of them at once, thus taking less time. This is what gives it its other name: massive parallel sequencing.
Despite the promise of next-generation sequencing, however, it’s yet to become a “conventional reality.” The design and workflow of NGS devices is very different from the normal DNA sequencers people are used to, which makes some people a bit suspicious of them. The usage of NGS in crime units is a pretty taboo concept right now, for instance — would you stake someone’s innocence on an experimental and unknown technology?
Of course, this may change sometime in the future.
Bioinformatics is the interdisciplinary combination of software tools to understand biological data, so technology is obviously involved somewhere along the way. Some of the key components in this field, as well as its purpose and recently developed examples, are software and devices. Whether it be to analyse an unidentifiable DNA sequence or remain as an archive for the sequences of specific people, software plays a significant role.
Think of Sanger sequencing. After the DNA fragments go through capillary electrophoresis, the obtained data will be useless and just shows several unlabelled graphs — unless machine learning steps in, to identify prominent features of chromatograms for example. There are several apps on the market that make this process more convenient.
The software in bioinformatics can also be used as a storage unit for sequences used for medical reasons. Let’s say you’re an oncologist who wants to conduct some research on the possible inhibition of leukaemia, and you’re not sure where to start. Luckily, there are several domains with growing numbers of DNA sequences of individuals with leukaemia, allowing you to perform research with much more ease. These “DNA hubs,” as I like to call them, are usually developed by large research facilities with similar goals as yours in mind.
Another key technological component is devices. Sequencing DNA manually is much too costly and time-consuming; biological processes that involve machinery are much more efficient. Machines are much better at processing and saving information, after all.
And that leaves humans to do what they do best: generate more DNA to sequence.